
We are broadly interested in applying machine learning towards problems in the following areas:
Genetics of human disease
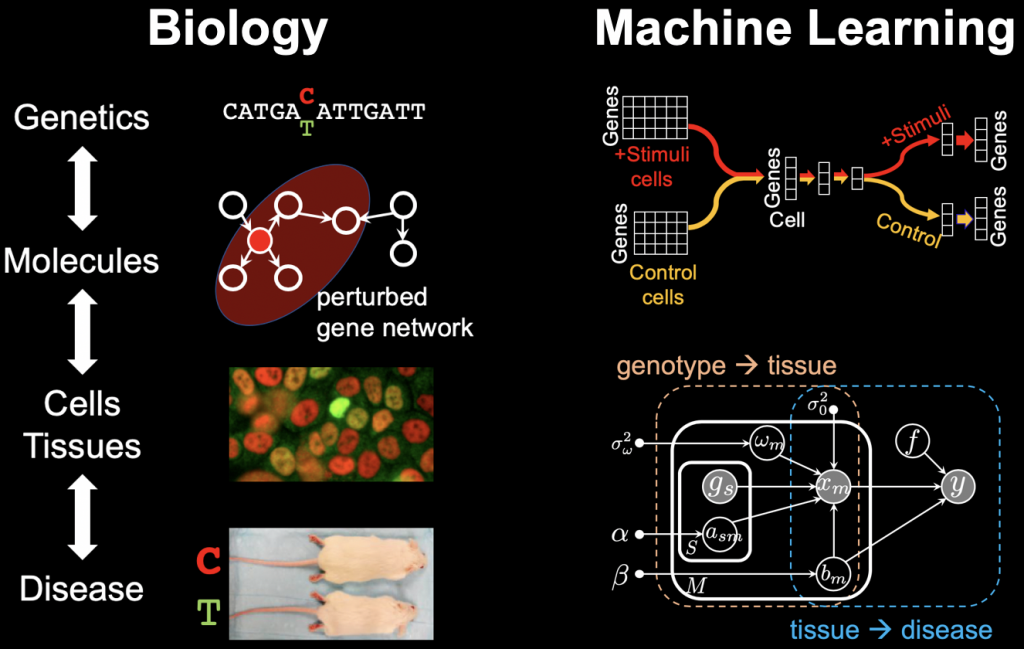
Several projects in the lab focus on building models to predict how genetic variation ultimately impacts disease risk through different layers of biological complexity, including molecules (e.g. gene regulation, gene expression, chromatin accessibility) and cell- and organ-level phenotypes measurable through live-cell imaging or MRIs. Our goals are two-fold: to understand the mechanism of action through which genetic variation modulates our risk of different diseases and traits, as well as to identify actionable therapeutic targets. We have published a number of papers on characterizing the genetics of obesity and Alzheimer’s disease, and have more recently focused on psychiatric and other mental disorders.
Models of cell population dynamics
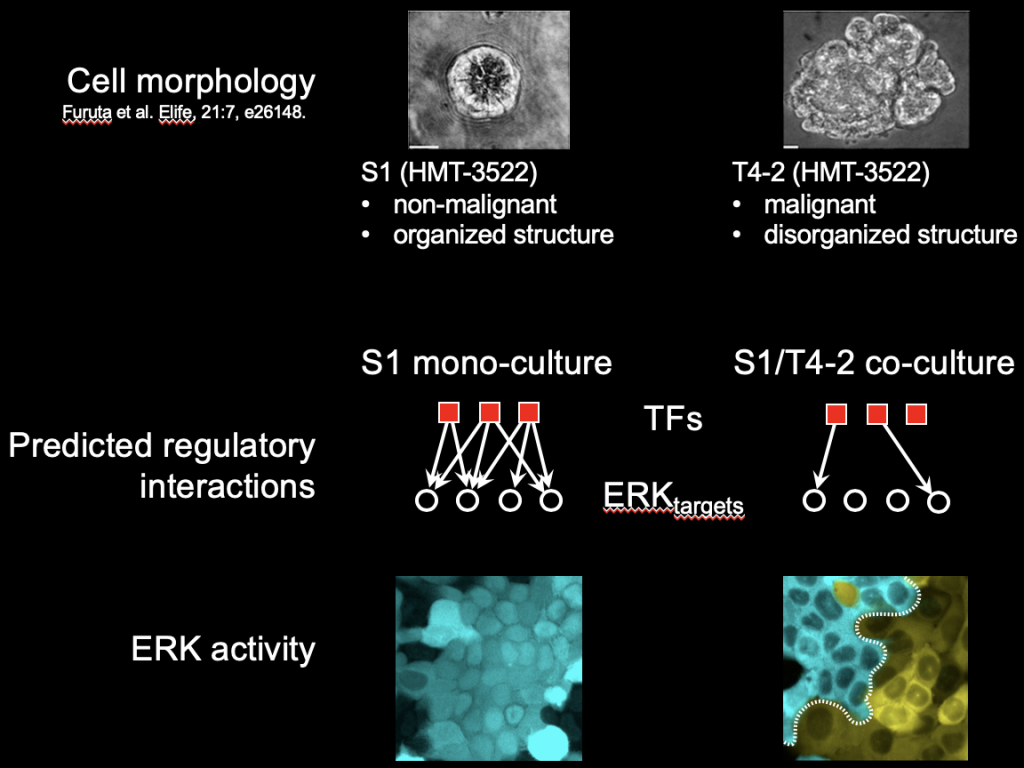
We are developing neural network-based models of population-level behavior of single cells. Sequencing technologies are routinely used to generate high resolution, high dimensional snapshots of individual cells. However, they historically are poor at capturing cell population-level behavior as well as time-varying behavior, because cells are sacrificed before sequencing. On the other hand, imaging-based technologies can be used to observe e.g. transcriptional behavior over time of a limited number of genes. Here we combine imaging-based and sequencing-based methodologies to model how cells interact and influence each other’s transcriptome. Shown in the figure above is an illustration of one such project, where we used single cell genomics data on non-malignant cells cultured either alone (S1) or co-cultured with malignant cells (S1 co-cultured with T4-2), to predict that S1 gene regulation of the ERK pathway is disrupted by proximity to T4-2 cells. This is evident both at the gene regulatory network level, as well as through reporter assays of individual genes.
Neurogenomics
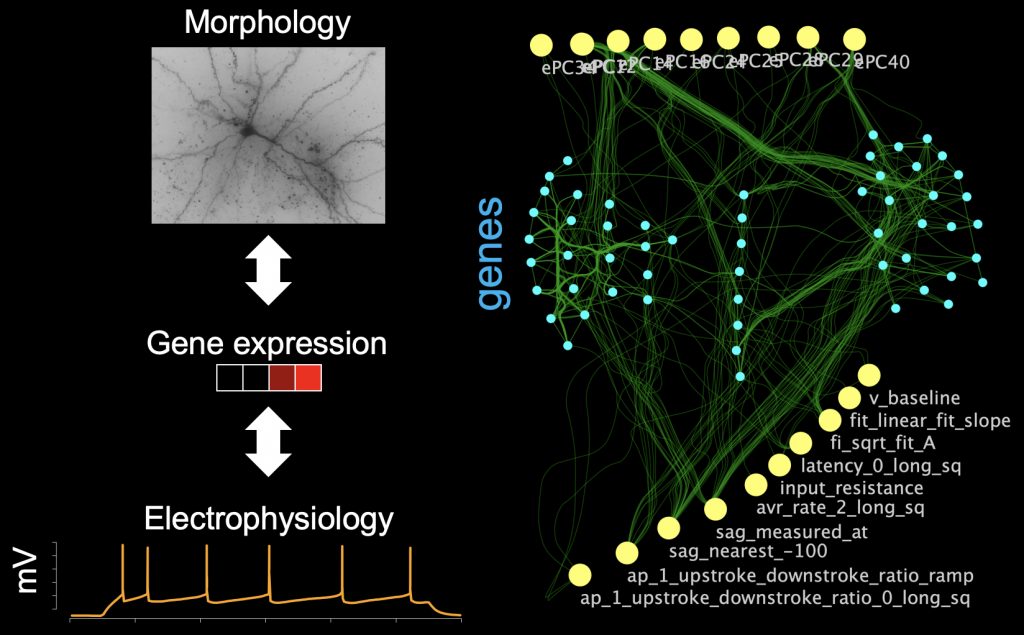
Our understanding of the dynamics of molecular events in the cell has been rapidly increasing, thanks to the many ways in which DNA sequencing has been repurposed to study different aspects of gene regulation. The relationship of gene regulation to cellular-level phenotypes and events, however, is poorly understood, in large part because they are hard to jointly measure experimentally. Several projects in the lab aim to bridge this gap by building quantitative neural network-based models to characterize how gene expression and electrophysiological/morphological properties of neurons co-vary, in order to then understand how changes in gene regulation caused by disease also change properties of neurons.